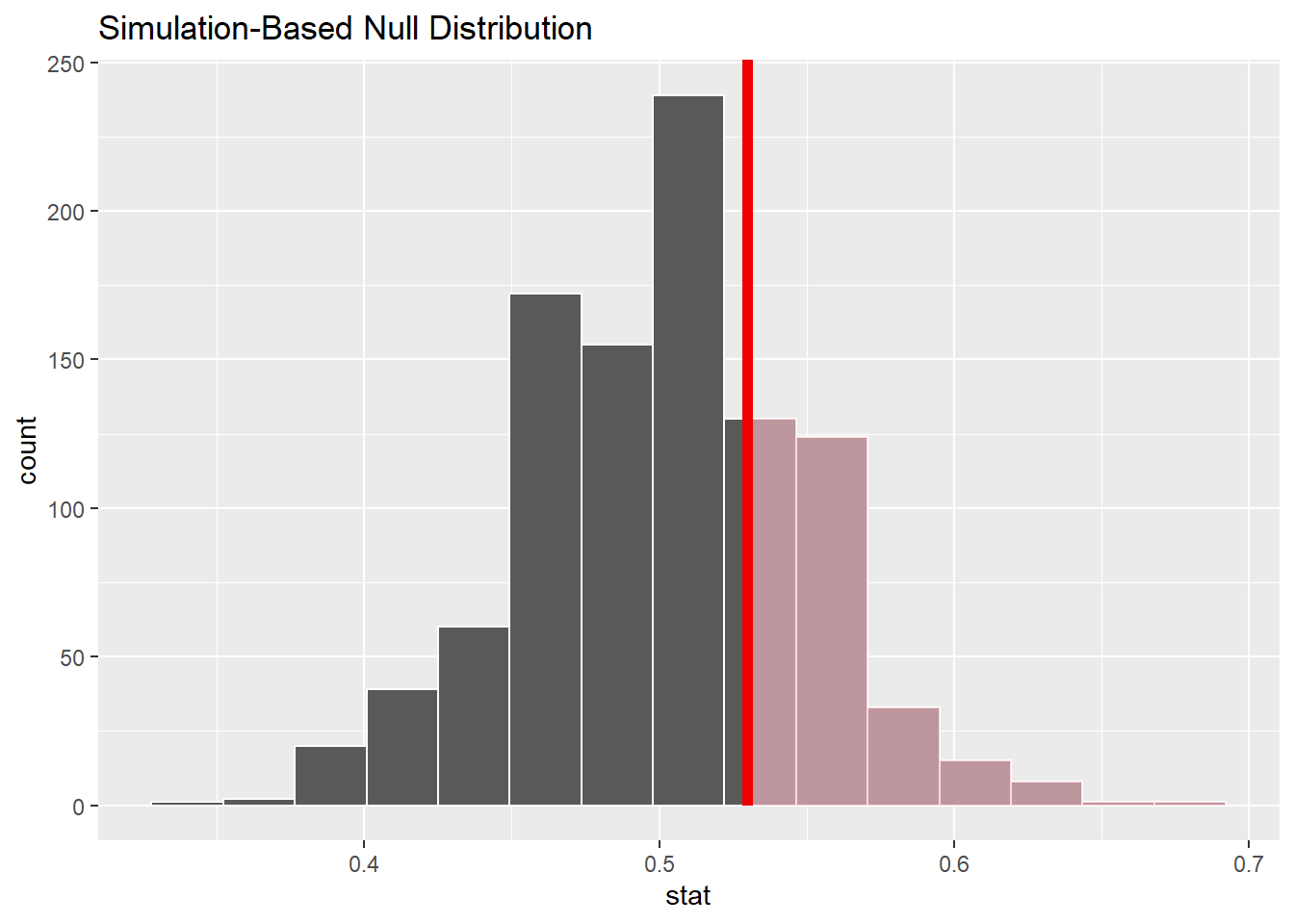
We know that all of you can read martian :)…. but let’s assume that another class that went through this study provided a sample statistic of \(\hat{p}\) = 53/100
set.seed(12345)class_data<-tibble( correct_guess =c((rep("Correct" , 53)), rep("Incorrect" , 47)))null_dist_demo<-class_data|>specify(response =correct_guess, success ="Correct")|>hypothesize(null ="point", p =.5)|>#fill in the blankgenerate(reps =1000, type ="draw")|>#fill in the blankcalculate(stat ="prop")#fill in the blankvisualize(null_dist_demo)+shade_p_value(0.53, direction ="right")#fill in the blank
null_dist_demo|>get_p_value(.53, direction ="right")#fill in the blank
# A tibble: 1 × 1
p_value
<dbl>
1 0.312
Let’s assume that now we incorrectly accept the null and say that the true proportion of people that can identify bumba correctly is equal to 0.5
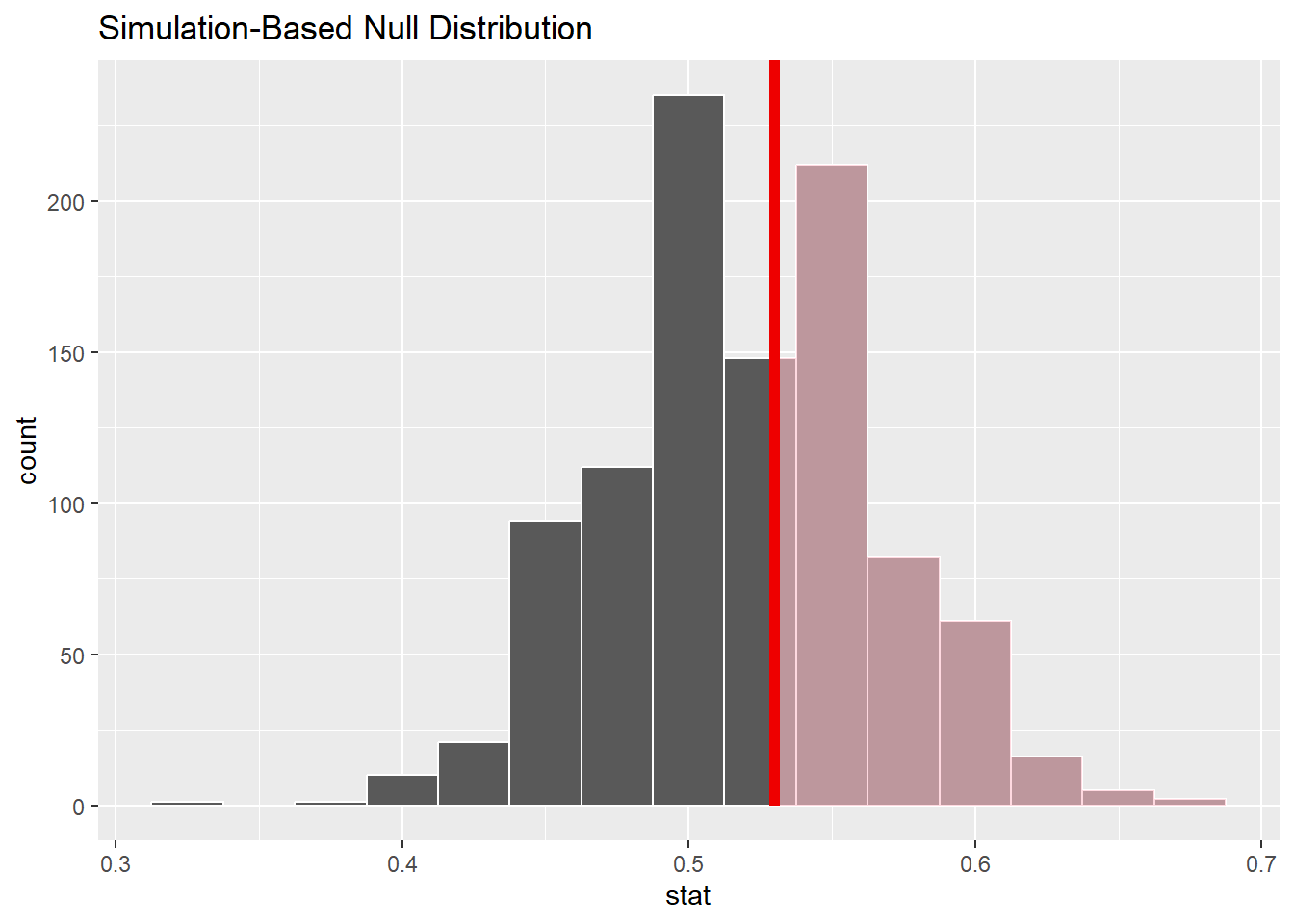
Assume that another researcher does the same study, calculated the same statistic (.53), but set up the null and alternative hypothesis as follows:
\(H_o: \pi = .52\)
\(H_a \pi > .52\)
set.seed(12345)class_data<-tibble( correct_guess =c((rep("Correct" , 53)), rep("Incorrect" , 47)))null_dist_demo2<-class_data|>specify(response =correct_guess, success ="Correct")|>hypothesize(null ="point", p =.52)|>#fill in the blankgenerate(reps =1000, type ="draw")|>#fill in the blankcalculate(stat ="prop")#fill in the blankvisualize(null_dist_demo2)+shade_p_value(0.53, direction ="right")#fill in the blank
null_dist_demo2|>get_p_value(.53, direction ="right")#fill in the blank
# A tibble: 1 × 1
p_value
<dbl>
1 0.456
This p-value is also very high…..
If we were to accept the null hypothesis, one researcher would say \(\pi = 0.5\) while the other would say \(\pi = 0.52\)…. and that doesn’t make sense!
This is why we use the language “fail to reject.”
More complex hypothesis test
Context
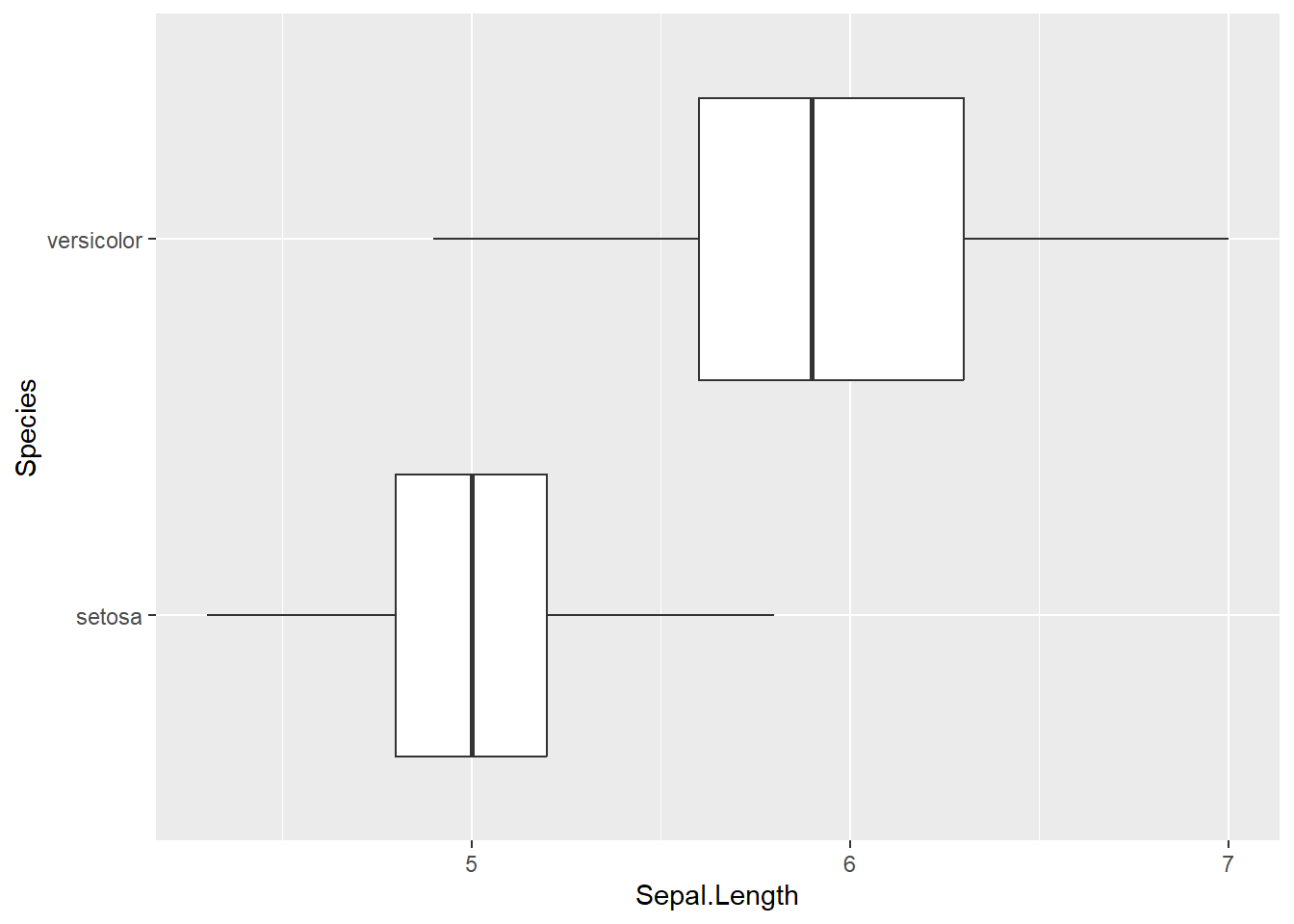
The Iris Dataset contains four features (length and width of sepals and petals) of 50 samples of three species of Iris (Iris setosa, Iris virginica and Iris versicolor). A sepal is the outer parts of the flower (often green and leaf-like) that enclose a developing bud. The petal are parts of a flower that are the pollen producing part of the flower that are often conspicuously colored. The difference between sepals and petals can be seen below.
The data were collected in 1936 at the Gaspé Peninsula, in Canada. For the first question of the exam, you will use this data sets to investigate a variety of relationships to learn more about each of these three flower species. The data set is prepackaged in R, and is called iris.
Now, we are going to see if this difference is by chance, or if this difference is meaningful…
Below, write out the null and alternative hypothesis in both words + notation.
\(H_0: \mu_{setosa} - \mu_{versicolor} = 0\)
\(H_a: \mu_{setosa} - \mu_{versicolor} \neq 0\)
Building a Distribution
Let’s use simulation-based methods to conduct the hypothesis test specified above. We’ll start by generating the null distribution. First, let’s start by calculating the sample size for each group.
# A tibble: 2 × 2
Species count
<fct> <int>
1 setosa 50
2 versicolor 50
Demo how 1 simulated difference in means in created
– PERMUTE or shuffle all observations together, regardless of their original species
– Distribute observations into two new groups of size n1 = 50 and size n2 = 50
– Calculate the new sample means for each group
– Subtract the new sample means
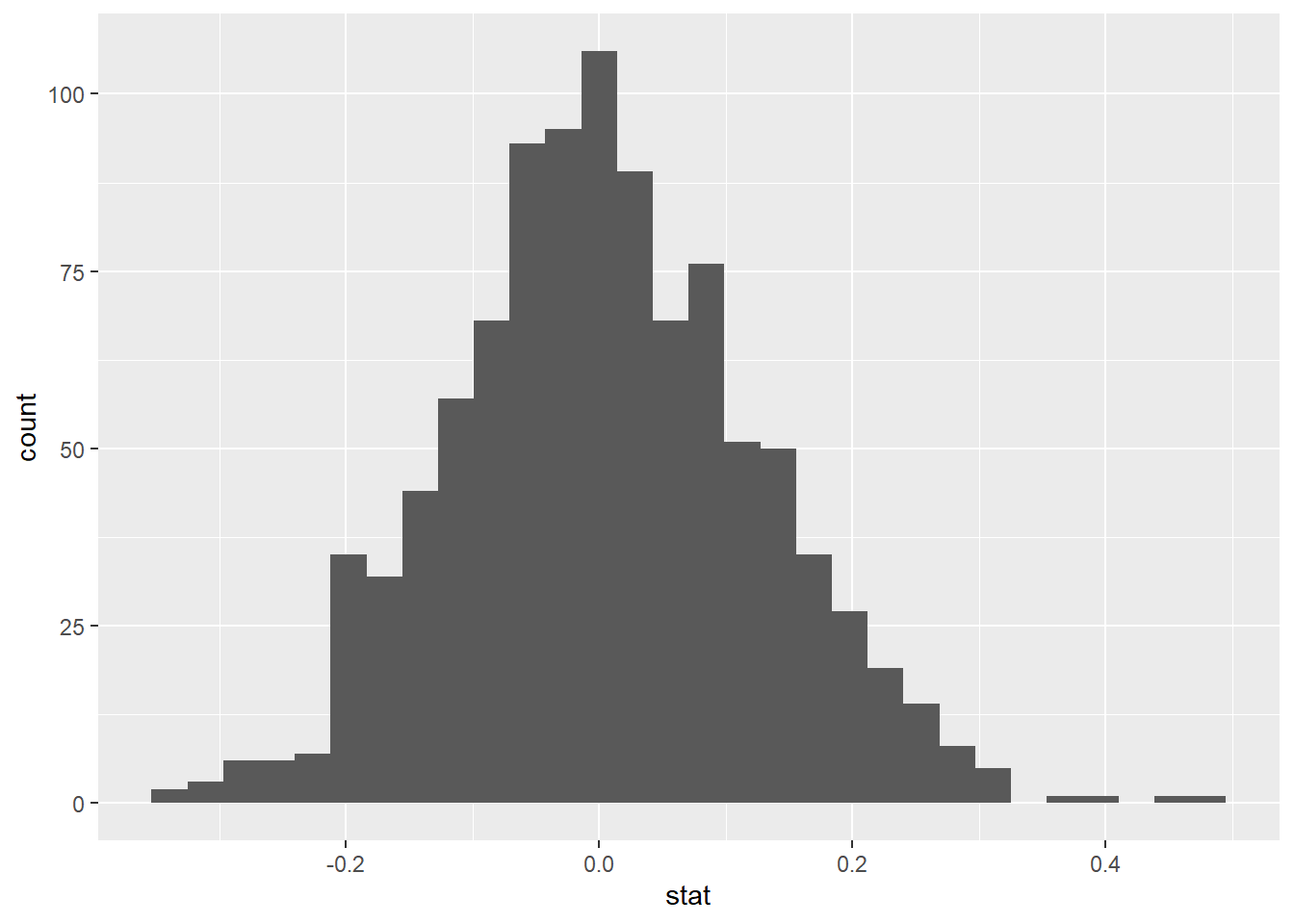
Now, create an appropriate visualization fo your null distribution using geom_histogram. Where is this distribution centered? Why does this make sense?
set.seed(12345)null_dist<-iris_filter|>specify(response =Sepal.Length, explanatory =Species)|>hypothesize(null ="independence")|>generate(reps =1000, type ="permute")|>calculate(stat ="diff in means", order =c("setosa", "versicolor"))
Dropping unused factor levels virginica from the supplied explanatory variable 'Species'.
`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.
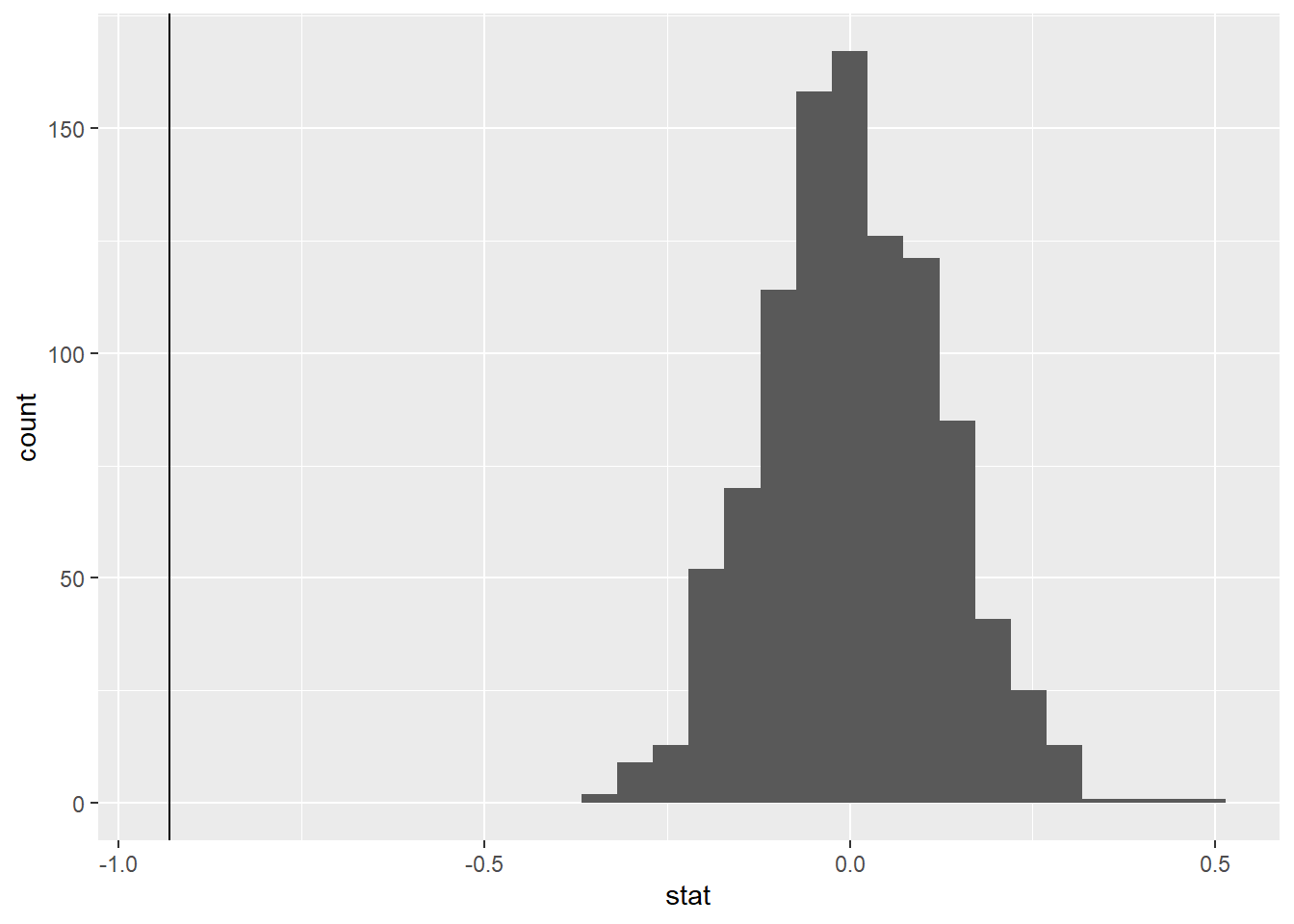
Now, use geom_vline to add a vertical line on your null distribution that represents your sample statistic. Based on the position of this line, do you your sample mean is an unusual observation under the assumption of the null hypothesis?
`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.
Calculate your p-value below
null_dist|>get_p_value(obs_stat =-0.93, direction ="two sided")
Warning: Please be cautious in reporting a p-value of 0. This result is an
approximation based on the number of `reps` chosen in the `generate()` step.
See `?get_p_value()` for more information.
# A tibble: 1 × 1
p_value
<dbl>
1 0
Write our an appropriate decision and conclusion in the context of the problem.
Decision: With a very small p-value, we reject the null hypothesis that the true mean sepal length for Setosa is equal to the true mean sepal length for Versicolor.
Conclusion: We have strong evidence to conclude that the true mean sepal length for Setosa is different than the true mean sepal length for Versicolor.